フェニルケトン尿症とは
三 フェニルケトン尿症(P.K.U.) これは常染色体劣性遺伝性代謝性疾患であり、10,000人に1人が罹患しており、ヘテロ接合体よりもホモ接合体で多く発生するようです。
高フェニルアラニン血症のグループに属するフェニルケトン尿症は、フェニルアラニンの代謝、特にその代謝を著しく損ないます への変換 チロシン;フェニルケトン尿症は、フェニルアラニンおよびいくつかの誘導体(フェニルピルビン酸、フェニル酢酸、フェニル酢酸およびフェニルアセチルグルタミン)の尿中レベルの上昇によって認識されます。
フェニルケトン尿症の最も深刻な合併症は 精神的な遅れ.
フェニルアラニン、チロシンおよび誘導体
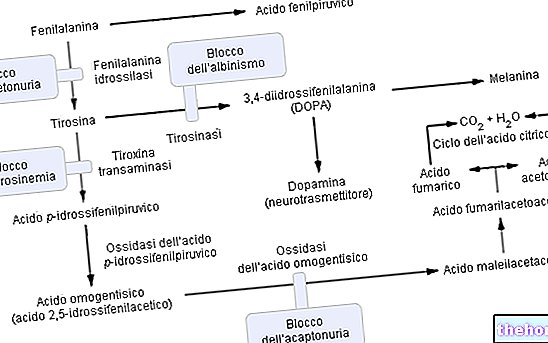
フェニルアラニンは必須アミノ酸であり、食事性タンパク質の大部分を構成しています。それは酵素によって変換することができます フェニルアラニンヒドロキシラーゼ チロシン中(ヒドロキシル基-OHを追加することにより)。次に、チロシンは以下の合成のための前駆体アミノ酸です:
- L-ドーパ(ドーパミン合成中間体)
- エピネフリン
- ノルエピネフリン(すべての神経伝達物質)。

フェニルケトン尿症のメカニズム(P.K.U.)
予想通り、フェニルケトン尿症では、1つまたは複数(全部で6つ)の染色体変異があるため、フェニルアラニンヒドロキシラーゼの発現(したがって代謝活性)は実質的にゼロです。これらの変化にはさまざまな種類があります(「ミスセンス」変化から「スプライシング」欠陥、さらには「部分的欠失」まで)が、重要なのは、この酵素の非効率性のために、ドミナントフェニルケトン尿症の血中フェニルアラニンレベル(通常は1mg / 100ml)です。それらは50倍も簡単に量に達します。
フェニルアラニンヒドロキシラーゼ酵素の機能:チロシン(+ジヒドロビオプテリン)を生成するために、フェニルアラニンヒドロキシラーゼは以下を必要とします:フェニルアラニン、酸素およびテトラヒドロビオプテリン(クーファクターとして作用する還元プテリジン);反応も可逆的であり、ジヒドロビオプテリンを再変換することができます(酵素のおかげで ジヒドロプテリンレダクターゼ)テトラヒドロビオプテリン中。
合併症
フェニルケトン尿症は、病理学的症状の重症度と診断の適時性に基づいて、多かれ少なかれ重篤な合併症を引き起こす可能性があります。遺伝性の病理であるフェニルケトン尿症は、次の点で区別されます。
- 優勢であるため、フェニルアラニンヒドロキシラーゼ酵素の完全な不活性を特徴とする
- 劣性、全酵素的遺伝の30%のみが活性である。
フェニルケトン尿症の合併症は、フェニルアラニン、その誘導体の代謝蓄積、およびチロシン合成の低下に起因し、直接比例します。病理学では、過剰なフェニルアラニンは、部分的にしか再吸収されない腎臓によって比較的効果的にろ過され、尿で排除されます。 ;しかし、高フェニルアラニン血症のレベルの持続性は、分子変換の代謝反応を決定します フェニルピルビン酸 および/または排出が容易な他の誘導体(フェニルピルビン酸、フェニル酢酸、フェニル酢酸)。
フェニルケトン尿症を複雑にしているのは、フェニルアラニン、フェニルピルビン酸、およびその誘導体の、中枢神経系(CNS)に対する毒性です。脳の発達におけるそれらの過剰な存在は、容赦なく精神遅滞の形態を決定します。
注意。他のアミノ酸の血漿中濃度は、おそらく腸管吸収または腎尿細管再吸収に関するフィードバックのために、わずかに減少しています。
フェニルケトン尿症の深刻な合併症としての脳損傷は、タンパク質合成、特にポリソーム、ミエリン、ノルアドレナリン、セロトニンの形成における他の必須アミノ酸の減算によって引き起こされます。フェニルケトン尿症-出生直後ではなく数年後には見えません-治療しない場合、子供の入院が必要であり、完全に不可逆的です。
進行したフェニルケトン尿症も肉眼ではっきりと見ることができます。高濃度のフェニルアラニン、酵素を阻害する チロシナーゼ、皮膚と髪の色素沈着を減らすことにより、メラニンの合成を著しく損ないます。さらに、髪や皮膚にフェニル酢酸が蓄積すると、フェニルケトン尿症患者に強くて不快な「マウスの臭い」が生じます。


















.jpg)