一般性
用語 網膜色素変性症 (RP)は、進行性網膜変性を特徴とする遺伝性疾患のグループを識別します。

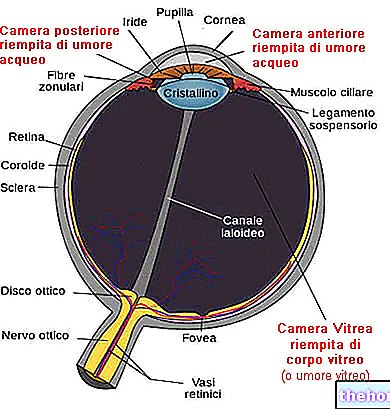
網膜色素変性症は、光受容体が徐々に失われ、色素上皮が機能不全になることを特徴とする網膜ジストロフィです。これは、網膜が視神経を介して視覚情報を脳に伝達する能力を徐々に低下させることを意味します。
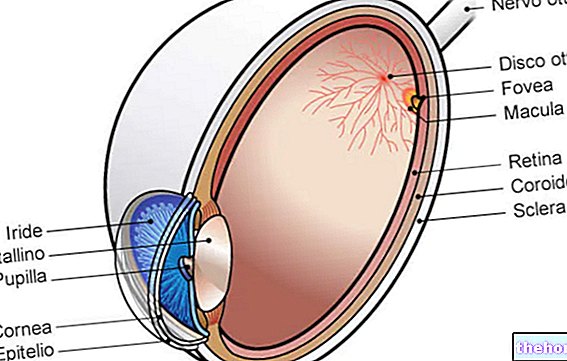
病理学的プロセスは、網膜色素上皮の変化から始まります。網膜色素変性症が進行すると、網膜に供給する血管が薄くなり、萎縮します。眼底を調べると、特徴的な沈着物が視覚的に検出されます。網膜色素(したがって、病気からの名前)。萎縮性の変化と損傷は視神経にも関係し、徐々に網膜の光感受性細胞が死にます。
網膜色素変性症に冒された患者は、特に照明が不十分な環境で最初に視力の問題を経験し、周辺視野の狭窄を訴えます。中心視力は病気の後期まで免れ、最終的な結果は劇的に変化する可能性があります。網膜色素変性症の多くの人は生涯を通じて限られた視力を維持しますが、他の人は完全に視力を失います。
網膜色素変性症は遺伝性疾患であり、主に片方または両方の親から受け継がれた遺伝的変化によって引き起こされます。遺伝的欠陥のタイプは、どの網膜細胞が障害に最も関与しているかを決定し、臨床的観点から、さまざまな状態を区別することを可能にします。現在までに、網膜色素変性症に関係する50以上の異なる遺伝的欠陥が確認されています。異常は、常染色体劣性、常染色体優性、またはヘテロソーム劣性(X連鎖またはX連鎖)の3つの遺伝パターンのいずれかを介して親から子孫に受け継がれる可能性があります。
症状
詳細情報:網膜色素変性症の症状
網膜色素変性症は通常、青年および若年成人に見られます。症状は10歳から30歳の間に現れることがよくありますが、診断は幼児期または人生のずっと後の段階で行うことができます。
網膜色素変性症の初期症状には以下が含まれます:
- 夜間(夜盲症)または暗い場所での視力の低下
- 暗闇での視力から明かりでの視力へ、またはその逆へのゆっくりとした適応。
- 視野の狭小化と周辺視野の喪失;
- 光とまぶしさに対する感度。
いくつかの症状は、関与する光受容体の種類によって異なります。桿体は黒と白の視力を担い、錐体は色を区別することができます。
網膜色素変性症のほとんどの場合、桿体が最初に関与します。ただし、急速に進化する形態では、コーンも初期段階で影響を受ける可能性があります。
桿体は網膜の外側に集中しており、薄暗い光によって活性化されるため、桿体の変性は末梢および暗視に影響を及ぼします。錐体が関与している場合、色覚と中心視力の喪失を経験する可能性があります。
関与する光受容体の優位性は、患者の遺伝子構成に存在する特定の欠陥によって決定されます。
多くの場合、網膜色素変性症の最初の症状は夜盲症(または夜盲症)です。一部の人々は、明るい場所から暗い場所に移動するにつれて、光の違いに適応するためにますます多くの時間が必要になることに気付きます。視力喪失の典型的な形態は、周辺視野(トンネルまたは望遠鏡の視力)の狭小化を引き起こします。このパターンはリング暗点と呼ばれます。時々、この現象は初期段階で見落とされるかもしれませんが、個人がしばしば物体につまずいたり、交通事故に巻き込まれたりするときに気づきます視力喪失が網膜の中心領域(黄斑ジストロフィーとも呼ばれます)患者を含む場合針の目を通して糸を通すなど、単一の物体に集中する必要がある読み取りや詳細な作業が困難になる多くの患者は、小さな、ちらつき、きらめく光として説明されることが多い光の閃光(光視症)を見たと報告しています。
病気の進行速度と視力低下の程度は人によって異なります。極端な場合には、20年以内に急速に進展するものもあれば、完全な失明につながることのない遅い経過もあります。早期発症は、より重症の網膜色素変性症に見られますが、軽度の状態(常染色体優性など)の患者は、生後50年または60年でこの疾患を発症する可能性があります。X染色体連鎖網膜色素変性症の家族では、男性がより頻繁に罹患します。一方、女性は遺伝的特徴を伝達し(X染色体上に変化した遺伝子を持っている)、障害の症状を示す頻度は低くなります。
合併症
網膜色素変性症は、ゆっくりではありますが、進行し続けます。ただし、完全な失明はまれですが、末梢および中枢の視力が大幅に低下する可能性があります。
網膜色素変性症の患者は、幼い頃に網膜の腫れ(黄斑浮腫)または白内障を発症することがよくあります。これらの合併症は、視力を妨げる場合に治療することができます。
関連疾患
一般的に、網膜色素変性症の患者には他の障害はなく、この場合、「非症候性」または単純な網膜色素変性症について話します。ただし、いくつかの症候群は、この眼疾患といくつかの臨床症状を共有しています。最も一般的なのはアッシャー症候群で、網膜色素変性症の全患者の約10〜30%に発症し、先天性または進行性の難聴を併発します。しかし、レーバー先天性黒内障では、子供は生後6か月以内に失明するか、ほぼ失明する可能性があります。網膜色素変性症に関連する他の疾患には、バルデービードル症候群やレフサム病などがあります。
原因
この病気は多くの遺伝的欠陥によって引き起こされる可能性があります:実際、変化の影響を受けた場合、網膜色素変性症の表現型を引き起こす可能性のあるいくつかの遺伝子があります。これらは通常、視力を可能にする伝達カスケードに関与するタンパク質をコードし、細胞の転写を促進します(網膜細胞に誤ったメッセージを送信する)または光受容体の構造を構成する要素の場合。遺伝性遺伝子変異は、受胎の瞬間から細胞に存在します。一般的な異常には、RP1遺伝子(網膜色素変性症-1、常染色体優性)の異常が含まれます。 、RHO(RP4、常染色体優性)およびRDS(RP7、常染色体優性)。網膜色素変性症の非遺伝性の原因はまれですが、それが存在しない孤立した症例(自然突然変異)を見つける可能性があります。病気。

.jpg)